
ライフサイエンス業界が迎える規制の転換期
ライフサイエンス業界は、最も複雑な規制環境の中で事業を展開しており、コンプライアンスと市場投入までのスピードが成功の鍵となります。
規制の枠組みが進化する中、企業は効率性と正確性を維持しつつ、複雑化する要件に的確に対応する必要があります。現在、製薬業界は規制申請のあり方そのものが変わる、大きな転換点にあります。 電子共通技術文書(eCTD)の最新バージョンであるv4.0は、申請プロセスの合理化とデータ交換の高度化を実現し、グローバルな規制申請の標準化と調和を目指しています。
eCTDとは?v4.0が重要な理由とは?
eCTD(Electronic Common Technical Document)は、医薬品の規制情報を各国の規制当局に提出するための標準フォーマットです。v4.0は、従来のeCTD v3.2を基盤としながら、構造、データ交換方法、効率性の面で大幅な改善を加えています。その目的は、世界中の規制申請を、より標準化・調和されたプロセスに導くことにあります。
eCTD v4.0の世界的な導入スケジュールと注目ポイント
世界各国の導入スケジュールは?なぜ日本とPMDAが注目されているのか?
導入スケジュールは地域によって異なります。
PMDA(日本):世界で初めて2026年からeCTD v4.0を義務化。2022年からパイロット申請を受け付けており、Freyrは2024年、2025年に予定されている初回申請に向けて、複数の日本企業を支援しています。
EMA(欧州医薬品庁):段階的な導入を進めており、一部の申請ではすでにv4.0が必要。今後すべての申請で義務化される予定です。
FDA(米国食品医薬品局):2029年の義務化を目指して準備中。
Health Canada(カナダ)は、2026年にオプションとしての申請を受け付け、2028年には必須となります。
TGA(オーストラリア)など他の地域当局も、それぞれ独自のスケジュールで導入を進めています。
任意提出の段階からスムーズに進めるためにも、規制当局が定める要件に早期から備えることが重要です。eCTD V4.0を早期に導入することで、必須化へのスムーズな移行が可能になります。各国の最新ガイダンスを常に把握し、適切な規制パートナーと連携することが、初回申請の成功に不可欠です。
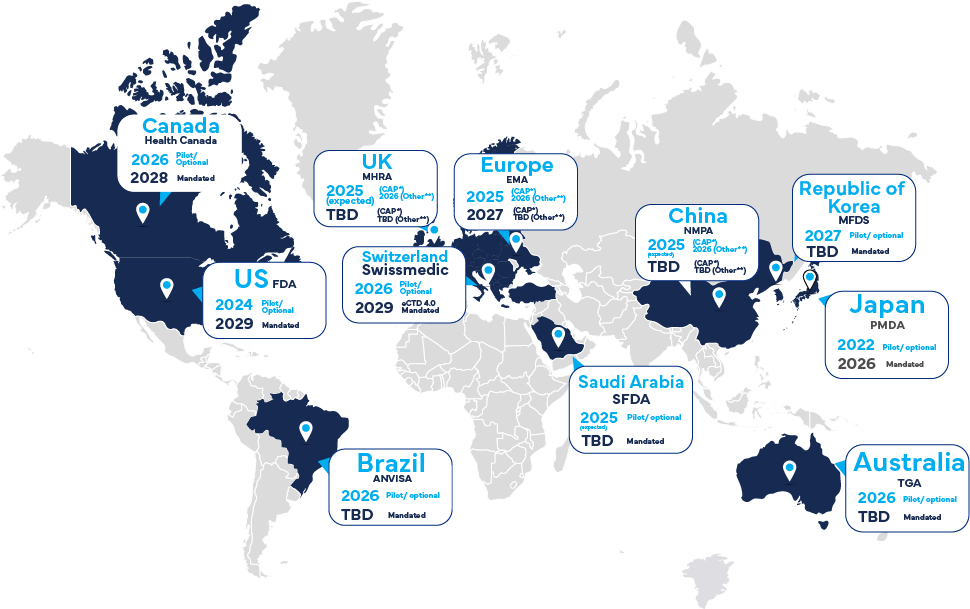
eCTD v4.0の申請受理は、米国FDAおよび日本PMDAですでに導入されており、他の主要な医療規制当局でも2025年〜2026年にかけて導入される予定です。
※CAPは中央承認手続(Centralized Authorization Procedure)を示します。
※「Other(その他)」は、MRP(相互認証手続)、DCP(分散手続)、NP(各国手続)を指します。
eCTD v4.0の主要な変更点と機能強化
PMDAにおけるeCTD v4.0は、従来のeCTD 3.2やJP 1.0とは大きく異なります。以下に主な変更点をまとめます:
モジュール1〜5を1つのXMLメッセージで統合。構造が簡素化され、一貫性が向上。
従来の「leaf(リーフ)」の概念は、「Context of Use(COU)」に置き換えられました。COUは、提出文書内での文書の目的とライフサイクルを定義します。これにより、製品のライフサイクル全体にわたって、文書の追跡および管理をより正確に行うことが可能になります。現在のプロセスでは、ファイルタグと階層構造に依存しています。
メタデータに標準化されたコントロールド・ボキャブラリ(CV)を使用することで、データの一貫性が向上。PMDAは日本独自のCV(JP CV)を開発し、地域要件への柔軟な対応を可能にしています。
eCTD v3.2.2との前方互換性を確保しており、過去の提出内容の参照・再利用が可能です。ただし、JP 1.0からの直接移行はサポートされておらず、別途準備が必要です。
eCTD v3.2.2とv4.0の比較
| 連番 | コンセプト | eCTD v3.2.2 | eCTD v4.0 |
|---|---|---|---|
| 1 | XML | 地域別 .xml | 単一のSubmission unit.xml |
| インデックス .xml | |||
| 2 | フォルダ構成 | 細分化されたフォルダ構造 | フラットなフォルダ構造 |
| 3 | メタデータ | ICH/地域別メタデータ | キーワードがeCTD v3.2.2の属性と値を置き換える |
| 4 | ライフサイクル管理 | ドキュメントのライフサイクル | 置換: |
| 置換:1対1 | 1対1 | ||
| 多対1 | |||
| 1対多 | |||
| 5 | ライフサイクル操作 | 新規 | 新規 |
| 追加 | 一時停止 | ||
| 置換 | 置換 | ||
| 削除 | |||
| 6 | シーケンス番号 | 0000または0001から開始 | 1から開始 |
| 7 | コンテンツの再利用 | 現在も可能だが運用が複雑で混乱を招きやすい | 各ドキュメントに固有識別子(UUID)が付与され、シーケンス間、規制活動間、異なる申請間でコンテンツの再利用が可能。 |
| ドキュメントにUUIDなし | (申請者は過去に提出したコンテンツを参照でき、新規シーケンスのサイズを削減可能。重複コンテンツの混乱も解消。UUIDはグローバルに受け入れられており、各当局間で真の調和が実現) | ||
| 8 | STF(スタディ・タグ付けファイル) | 各治験ごとに単一XML | モジュール4および5のContext of UseがSTFを置き換える |
| 9 | ドキュメントの順序 | 現在は各レビューツールで定義されており、ツール間で異なる場合がある | eCTD v4.0では、申請者が特定セクション内のファイルの表示順序を明示的に定義可能 |
| 10 | キーワード/属性の修正 | eCTD属性(例:製造者名)のわずかな表記揺れでも別のeCTDセクションが作成される可能性あり | eCTD v4.0では、申請者が定義するキーワード/属性を管理でき、誤りや変更の修正が容易 |
eCTD v4.0の導入による戦略的メリット
また、現行の v3.2.2 と比較した場合の eCTD v4.0 の主な利点 を以下にご紹介します。
Key Advantages
| 番号 | 主な利点 | 詳細 |
|---|---|---|
| 1 | 提出単位の統一 | •モジュール1からモジュール5までのすべての内容が、1つの交換メッセージに含まれます。 つまり、1つのXMLファイルでICH情報と地域情報の両方をカバーします。 |
| 2 | ドキュメントの再利用 | •一度提出された文書は、同一または異なる申請で一意の識別子(ID)を参照することで再利用可能 |
| •再利用されるドキュメントのすべての内容(他のドキュメントへの参照やハイパーテキストリンクを含む)は、再利用先の申請に関連している必要がある。 | ||
| 3 | コンテキスト・オブ・ユース(COU)(Context of Use) | •CTD見出し+ドキュメント+キーワードで構成。 |
| •コンテキスト・オブ・ユース(COU)の概念により、より高度なライフサイクル管理操作が可能となります。COUは、ライフサイクル管理を通じて、1つのCOUを複数のCOU要素に置き換えたり、その逆(一対多、多対一)も可能です。 | ||
| •eCTD v4.0では、物理ファイルやCOU要素自体を再提出することなく、キーワード定義の表示名(例:原薬/製剤名、製造者、剤形、適応症、添加剤、グループタイトルなど)を変更できる機能も導入されています。 | ||
| 4 | グループタイトルキーワードGroup Title Keywords | •グループタイトルキーワードはeCTD v4.0で新たに導入されたもので、申請者が定義するキーワードの一種です。グループタイトル・キーワードを用いることで、1つの見出しの下に複数の文書をグループ化して整理することが可能です。 |
| •グループタイトルに関連付けられたドキュメントは、eCTD見出しや他の関連キーワードとは別に表示されます。グループタイトル・キーワードは、最も下位の見出しレベルに適用されます。 | ||
| 5 | ドキュメントの表示順制御 | •CTDセクション内のドキュメントの順序を設定します。 |
| •コンテキスト・オブ・ユース(CoU)やコンテキストグループの下で、ドキュメントの表示順序を明示的に定義します。 | ||
| •申請者は、提出内容の順序を変更したり、既存の内容の中で特定の順序に新たな提出内容を挿入したりすることができます。 | ||
| 6 | ドキュメント識別子 | •すべてのドキュメントには固有の識別子が割り当てられます。 |
| •この固有識別子の仕組みにより、物理ファイルを再提出することなく、ドキュメントをより効果的に参照・再利用できるようになります。 | ||
| ・提出ユニット(シーケンス)内 | ||
| ・同一申請内の異なる規制活動間 | ||
| ・異なる申請間 | ||
| •コンテキスト・オブ・ユースやコンテキストグループ内でドキュメントを参照する際に使用されます。 | ||
| •ドキュメントのメタデータ(例:ドキュメントタイトル、場所)も再利用されます。 | ||
| 7 | v3.2.2からv4.0への互換性 | •v3.2.2からv4.0への前方互換性(移行用XMLの利用) |
| 8 | コントロールド・ボキャブラリ (CV:管理用語集) |
•5種類の語彙が使用可能 ・ICHが定める語彙 ・各地域が定める語彙 ・HL7による語彙 ・外部機関による語彙 ・送信者定義語彙 |
FreyrによるeCTD v4.0申請支援のご案内
次回のブログでは、eCTD v4.0申請の具体的な流れや地域別の違い、日本・欧州・米国における申請プロセスの詳細をご紹介します。
Freyrは、日本・欧州・米国において、多くのクライアントのeCTD v4.0ライブ申請およびパイロット申請を支援してきた豊富な実績があります。PMDA、FDA、EMAと密接に連携し、薬事申請の効率化と医薬品の迅速な市場投入を支援しています。
eCTD v4.0への円滑な移行や、より効率的な申請プロセスをご希望の方は、Freyrまでご相談ください。

著者について
ラガヴェンドラン・バブ(ラガヴ)氏は、グローバルな保健当局への申請業務において20年以上の経験を持つ、熟練した規制業務のリーダーです。Freyr社の規制業務ディレクターとして、規制申請戦略、eCTD/NeeS/紙媒体での申請、さらにはAIの応用や構造化コンテンツソリューションなどの革新的な規制テクノロジーにも注力しています。
業界における専門家として、eCTD v4.0、SPL、SPM、ePIなど進化する規制標準を主要市場で先導的に導入・実装した実績があります。技術チームと密接に協働し、規制ビジネスのニーズと実用的かつ拡張可能なソリューションとの整合を図る一方で、社内外のトレーニングプログラムも主導し、規制対応力の強化にも貢献しています。ラガヴ氏は、化学の学士号およびシステム&情報技術専攻の経営学修士号(MBA)を取得しています。

モヒト・バトラ博士は、医学博士であり、ライフサイエンス、デジタルヘルス、規制業務分野で12年以上にわたるグローバルな経験を有する優れた戦略コンサルタントです。現在はFreyr Solutionsにて、規制業務のグローバルビジネスおよび戦略部門の責任者(シニアディレクター)を務めており、医学的専門知識と戦略的洞察を融合させた独自の視点を提供しています。バトラ博士は、デリー大学で医学博士号(MD)を取得し、インド経営大学院(Indian School of Business)でMBAを取得しています。