
The Life Sciences Industry Faces a Regulatory Turning Point
The life sciences industry operates in one of the most complex regulatory environments, where compliance and speed-to-market are critical for success.
As regulatory frameworks evolve, organizations must navigate increasing complexities while ensuring efficiency and accuracy. At present, the pharmaceutical industry stands at the cusp of a significant shift in regulatory submissions. eCTD v4.0, the latest evolution of the electronic Common Technical Document, promises to streamline processes and enhance data exchange.
What Is eCTD and Why Is Version 4.0 So Important?
Electronic Common Technical Document (which we commonly refer to as eCTD) refers to the standard format for submitting regulatory information for medicinal products, across any country globally. eCTD v4.0 builds upon the foundation of the previous version of eCTD 3.2 but introduces significant enhancements in terms of structure, how data is exchanged between relevant parties, and overall efficiency. That key goal is for a more standardized and harmonized approach to regulatory submissions worldwide going forward.
Global Timelines for eCTD v4.0 Implementation
What are the global implementation timelines for eCTD v4.0? Also, why is Japan and PMDA the current focus of eCTD v4.0 discussions?
The implementation timelines vary for different regions.
The PMDA (Japan) is the first agency globally to make eCTD v4.0 mandatory from 2026 and they have already started accepting pilot submissions from 2022. Interestingly, Freyr is helping multiple clients in Japan for their first 4.0 submission in 2024 and 2025. We will talk about it a little later.
The EMA (European Medicines Agency) has a phased plan with certain procedures already requiring eCTD v4.0, aiming for mandatory adoption for all applications in the coming years.
The FDA (U.S. Food and Drug Administration) is also progressing with its plans, aiming for 2029 for mandatory submissions.
The Health Canada (HC) In 2026, Health Canada will accept optional submissions; in 2028, they will be mandatory.
Other regions like Australia's TGA Other regions like Australia's TGA are also working towards adoption with their own timelines.
Learn how to facilitate optional submissions and prepare for the mandatory requirements set forth by the health authority. Early eCTD V4.0 adoption facilitates a smooth transition to required adoption.
Hence, it is of utmost importance for companies to stay updated on each relevant authority's specific guidance and work with the right regulatory partner for successful submissions in the first go.
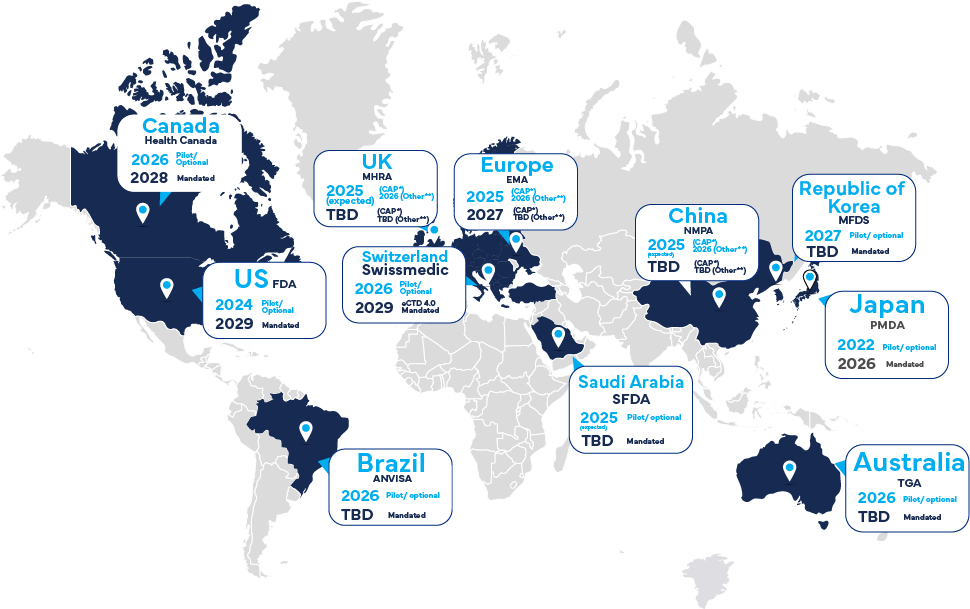
Acceptance of eCTD v4.0 submissions has been implemented by US FDA and Japan PMDA and will be implemented by other major health agencies in 2025/2026.
*CAP indicates Centralized Authorization Procedure.
** Other indicates MRP (Mutual Recognition Procedure), DCP (Decentralized Procedure) and NP (National Procedure)
What's New in eCTD v4.0 - Major Shifts and Enhancements
eCTD v4.0 brings many changes for PMDA and differs considerably from the current processes namely eCTD 3.2 and JP 1.0. Below we are highlighting some of the key changes
Unlike the previous eCTD versions, eCTD v4.0 uses a single XML message to encompass Modules 1 through 5. This simplifies the structure and aims for greater consistency. Currently, PMDA deals with regional-specific XML structures in Module 1 alongside the ICH XML Modules 2-5.
The concept of a "leaf" is replaced by "Context of Use." COU defines the purpose and lifecycle of a document within the submission. This allows for more precise tracking and management of documents throughout the product's lifecycle. The current process relies on file tags and a hierarchical structure.
eCTD v4.0 mandates the use of standardized controlled vocabularies for metadata. PMDA has developed a Japan-specific controlled vocabulary (JP CV) to capture local nuances. This will improve data consistency and facilitate easier updates to regional requirements without major system changes.
eCTD v4.0 is designed to be forward compatible with eCTD 3.2.2, allowing for the referencing and potential reuse of previously submitted content. However, there is no direct migration path from the older Japan-specific JP 1.0 format to eCTD v4.0, requiring a more significant preparation effort for submissions that were initially in that format.
Comparison - eCTD v3.2 vs eCTD v4.0
| S. No. | Concept | eCTD v3.2 | eCTD v4.0 |
|---|---|---|---|
| 1 | XML | Regional .xml Index .xml | Single Submission unit.xml |
| 2 | Folder Structure | Granular Folder Structure | Flat Folder Structure |
| 3 | Metadata data | ICH/ Regional Metadata | Keywords replace the eCTD 3.2.2 attributes and valid value |
| 4 | Lifecycle |
Document Life cycle Replace:
|
Replace:
|
| 5 | Life cycle Operations | New Append Replace Delete | New Suspend Replace |
| 6 | Sequence Number | Starts from 0000 or 0001 | Sequence number starts from 1 |
| 7 | Reuse of Content |
Currently Possible but Difficult and Confusing No UUID for documents |
Unique Identifiers (UUID) for each document allow reuse of content across sequences, regulatory activities and different applications. (Sponsors may refer to previously submitted content and reduce the size of new sequences. Confusion over duplicate content is removed. Since the UUID is accepted globally, true harmonized submissions across agencies will become a reality) |
| 8 | STF (Study Tagging File) | Single XML for each clinical studies | Context of Use in Module 4 and Module 5 will replace the STF |
| 9 | Document Ordering | Currently defined by each review tool; may not be the same between tools | eCTD 4.0 allows the submitter to explicitly define the display order for files in a specific section |
| 10 | Keyword/ attribute modifications |
Currently a slight misspelling in eCTD attributes (e.g., manufacturer) creates separate eCTD sections |
eCTD 4.0 user-defined keywords/ attributes are managed by the submitter and mistakes/ changes are easier to correct |
Strategic Advantages of eCTD v4.0
Also, we are listing some of the key advantages of eCTD v4.0 over the current v3.2.2
Key Advantages
| S. No. | Key Advantage | Details |
|---|---|---|
| 1 | Harmonized submission unit |
|
| 2 | Document reuse |
|
| 3 | Context of Use (COU) |
|
| 4 | Group Title Keywords |
|
| 5 | Harmonized submission unit |
|
| 6 | Document Identifier |
|
| 7 | Forward Compatibility from v3.2. to v4.0 |
|
| 8 | Controlled vocabularies (CV) |
|
How Freyr Can Help
Ready to streamline your regulatory submissions? In our next blog, we're pulling back the curtain on eCTD v4.0. We'll walk you through what a typical v4.0 submission looks like, uncover the critical regional variations, and share expert insights into navigating the distinct processes for Japan, the EU, and the US.
Freyr is already a trusted partner for many clients in Japan, the US, and the EU, guiding them through their live and pilot eCTD v4.0 submissions. By working hand-in-hand with the JP PMDA, US FDA, and EMA, we're committed to helping you accelerate the delivery of vital medicines to patients worldwide.
Curious about how we can help you achieve faster, more efficient submissions? Reach out to Freyr today!

About the Authors
Ragavendran Babu (Ragav) is a seasoned regulatory operations leader with over 20 years of experience across global health authority submissions. As Director of Regulatory Operations at Freyr, he specializes in regulatory submission strategy, eCTD/NeeS/Paper submissions, and emerging innovations in regulatory technology, including AI applications and structured content solutions.
Ragav has served as an industry subject matter expert, successfully piloting and implementing evolving regulatory standards such as eCTD v4.0, SPL, SPM, and ePI across major global markets. He works closely with technology teams to align regulatory business needs with practical, scalable solutions, while also leading internal and external training programs to build regulatory readiness.
He holds a Bachelor’s degree in Chemistry and a Master’s in Business Management (Systems & Information Technology).

Dr. Mohit Batra is a medical doctor and an accomplished strategy consultant with over 12 years of global experience in life sciences, digital health, and regulatory affairs. As Senior Director and Global Business and Strategy Head of Regulatory Operations at Freyr Solutions, he brings a unique blend of medical expertise and strategic insight. Dr. Batra holds an MD from Delhi University and an MBA from the Indian School of Business.