
当ブログはeCTD v4.0進化に関するシリーズの第2弾で、典型的なeCTD v4.0提出の流れや地域ごとに異なるステップ、日本PMDA向け提出プロセスの重要な知見を中心に解説します。
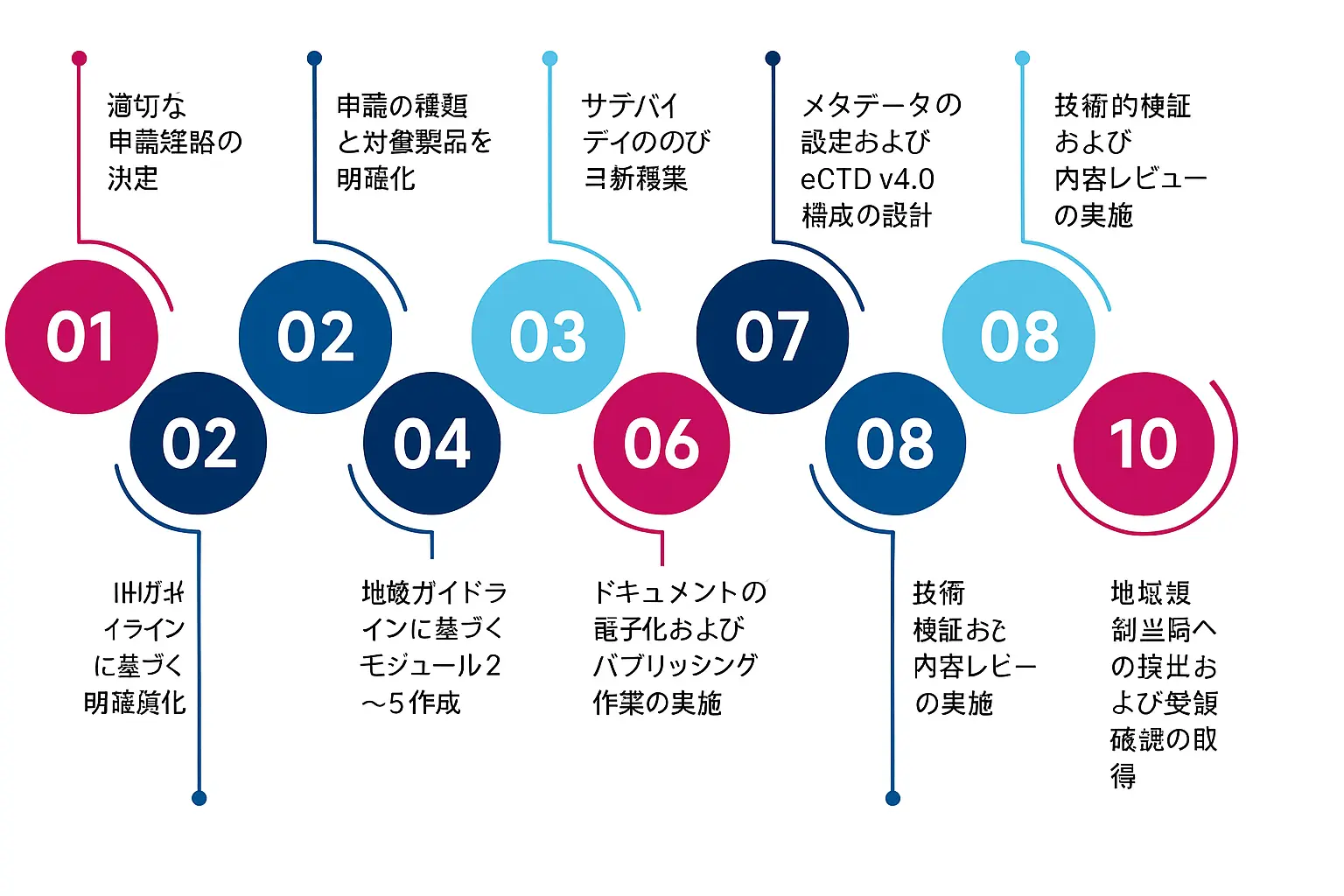
典型的なeCTD提出プロセスは、適切な規制ルートや提出タイプの決定、モジュール1から5の準備、編纂、品質管理、検証から最終提出まで多くのステップを含みます。しかし、各地域は独自の要件を持っているため、不要な遅延を避けるには適切な専門家との連携が必須です。ここでは、代表的な世界の提出プロセスと日本・EU・米国の主な違いを紹介します。
eCTD v4.0 の申請プロセス(例)
提出プロセス:日本vs 欧州 vs 米国
| Japan | EMA | US FDA |
|---|---|---|
|
2通りの提出方式
|
手続きの種類に基づく4段階プロセス
|
2ステップ方式
|
PMDAのeCTD v4.0導入について知っておくべきこと
ここで日本について詳しく見ていきましょう。日本の現在の規制提出プロセスは主に電子化されていますが、一部の場合には紙媒体での提出も認められています。医薬品医療機器総合機構(PMDA)は、これらの提出書類の審査および承認を担当する中央機関です。
それだけでなく、前述した日本、米国、EUの主な違いで説明したように、日本では初回NDA申請において適切な提出タイプを特定し、選択したカテゴリーに基づいてPMDAへの提出スケジュールを策定する必要があります。以下に示す通りです。
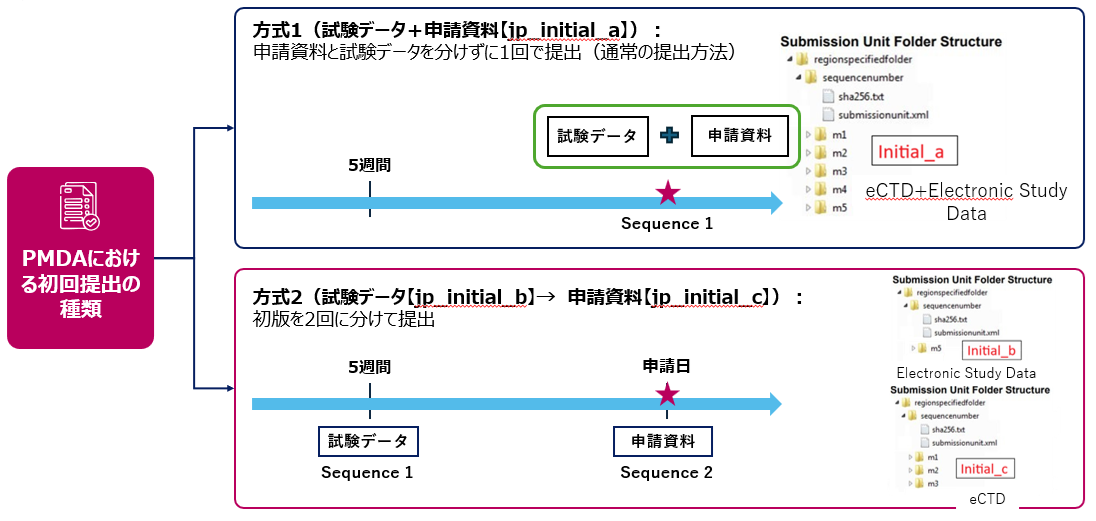
初版提出時における当該Submission Unitの種類
eCTD Initial NDA submission Types
試験データ+申請資料
初版提出時に、
CTD通知によって定められた
資料及び申請電子データを、
一つのeCTD v4.0
XML メッセージから
参照して提出するeCTD。
試験データ
初版提出時に、申請電子データのみを一つのeCTD v4.0 XML メッセージインスタンスから
参照して提出するeCTD。
申請資料
初版提出時に、
CTD通知によって
定められた資料のみを
一つのeCTD v4.0 XML メッセージインスタンスから
参照して提出するeCTD。
jp_other
対応可能な手段が他に無くやむを得ない理由がある場合に限り使用する。使用にあたっては事前に審査当局に相談すること。
PMDA eCTD v4.0:方式
古いフォーマットでの提出はどうなる?eCTD v4.0への移行は可能か?
PMDAおよび他の保健当局におけるeCTD v4.0の義務化実施日以降は、非eCTD v4.0フォーマットでの申請提出が却下されたり、再提出を求められたり、審査に遅延が生じる可能性があります。
しかしながら、eCTD v4.0の構造は、従来のフォーマットで提出された書類の再利用を可能としている点は朗報です。ただし、移行に伴う作業のプロセスや負荷は、複数の要因によって異なります。
紙ベースの提出や非常に古い電子フォーマット(例えばNeeS: Non-eCTD electronic Submission)からの移行は、eCTD 3.2.2からの移行に比べて大幅に複雑になります。
構造が整備された既存のドシエは、eCTD v4.0の構造への対応が容易です。
eCTD v4.0に関する専門知識を有する人材および専用ソフトウェアは、円滑な移行を成功させる上で不可欠です。
保健当局によっては、移行支援のためのガイダンスやツールを提供する場合もありますが、申請者に一任する場合もあります。
eCTD v4.0への移行における重要なステップ
過去の提出書類を移行するには、eCTD v4.0に関する十分な知識と適切な実行プロセスが必要です。主なステップは以下の通りです。
ギャップ分析
古いドシエの構造や内容をeCTD v4.0の要件と比較し、データや形式上の不足点を明確に特定します。
文書の再編成
既存文書をモジュール構造およびeCTD v4.0のより細分化された体系に合わせて再編成します。
メタデータの更新
全文書のメタデータをeCTD v4.0の仕様に準拠するよう更新します。これには制御語彙(コントロールド・ボキャブラリ)やUUID(Universally Unique Identifier)などの新しいデータ要素が含まれます。
XMLバックボーンファイルの生成
ドシエの構造を正確に反映し、文書と対応メタデータをリンクさせたeCTD v4.0用のXMLバックボーンファイルを作成します。eCTD v4.0では従来の複数ファイル形式から、単一XMLファイルに統合されています。
技術的検証
適切なソフトウェアを使用して新たに作成したeCTD v4.0提出物の技術的適合性を検証し、保健当局の仕様に準拠していることを確認します。
提出
新フォーマット化されたeCTD v4.0ドシエを保健当局へ提出します。
したがって、eCTD v4.0提出をスムーズに進めるためには、過去の提出物も含めて確かな実績を持つパートナーとしっかり連携することが、成功の鍵となります。
規制提出の効率化にご関心はありませんか?次回のブログでは、制御語彙の地域差や最新規制に対応したツールの重要性、さらに日本、米国、EUのクライアントや保健当局との豊富な経験に基づくベストプラクティスについて詳しくご紹介します。
より迅速で効率的な提出を目指すなら、ぜひFreyrにご相談ください。お客様のニーズに合わせたサポートで、スムーズな申請プロセスを実現いたします。 お気軽にFreyrまでお問い合わせください!

著者について
ラガヴェンドラン・バブ(ラガヴ)氏は、グローバルな保健当局への申請業務において20年以上の経験を持つ、熟練した規制業務のリーダーです。Freyr社の規制業務ディレクターとして、規制申請戦略、eCTD/NeeS/紙媒体での申請、さらにはAIの応用や構造化コンテンツソリューションなどの革新的な規制テクノロジーにも注力しています。
業界における専門家として、eCTD v4.0、SPL、SPM、ePIなど進化する規制標準を主要市場で先導的に導入・実装した実績があります。技術チームと密接に協働し、規制ビジネスのニーズと実用的かつ拡張可能なソリューションとの整合を図る一方で、社内外のトレーニングプログラムも主導し、規制対応力の強化にも貢献しています。ラガヴ氏は、化学の学士号およびシステム&情報技術専攻の経営学修士号(MBA)を取得しています。

モヒト・バトラ博士は、医学博士であり、ライフサイエンス、デジタルヘルス、規制業務分野で12年以上にわたるグローバルな経験を有する優れた戦略コンサルタントです。現在はFreyr Solutionsにて、規制業務のグローバルビジネスおよび戦略部門の責任者(シニアディレクター)を務めており、医学的専門知識と戦略的洞察を融合させた独自の視点を提供しています。バトラ博士は、デリー大学で医学博士号(MD)を取得し、インド経営大学院(Indian School of Business)でMBAを取得しています。