
In our blog 2 of the series about eCTD v4.0 evolution, we will focus on a how a typical eCTD v4.0 submission looks like, how some steps differ for different regions and share key insights on the submission process for Japan PMDA.
A typical eCTD submission process involves many steps from determining the right regulatory pathway and submission type, preparing Modules 1 to 5, compiling, QC, validating to final submission. But each region has its own specific requirements, which necessities partnering with the right experts to avoid any preventable delays. We are highlighting a typical global submission process and the key differences between Japan, EU and US.
eCTD v4.0 Submission Process
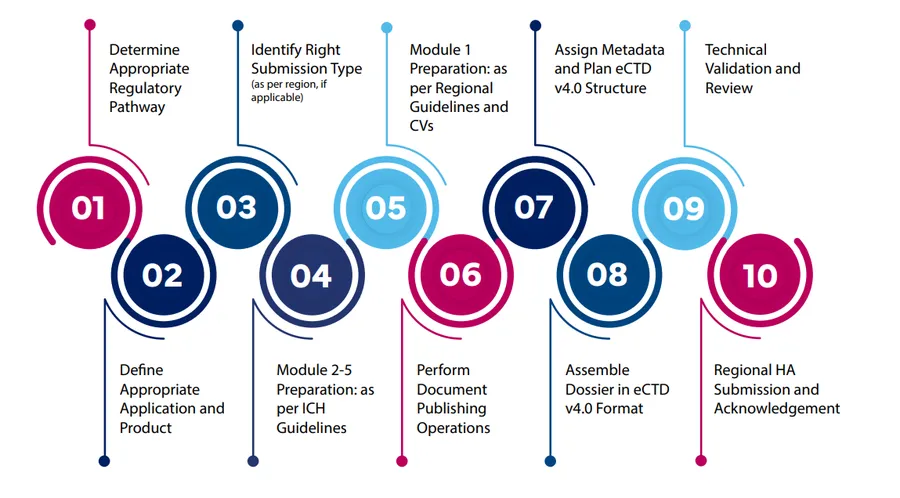
Submission Process: JP vs US vs EU
| Japan | EMA | US FDA |
|---|---|---|
|
2 Way submission Process
|
Based on Procedure types
|
2 step process
|
What You Need to Know About PMDA's eCTD v4.0 Adoption
Now let's deep dive into Japan. The current regulatory submission process in Japan is primarily electronic but with acceptance of paper submission in some instances. The Pharmaceuticals and Medical Devices Agency (PMDA) is the central authority responsible for reviewing and approving these submissions.
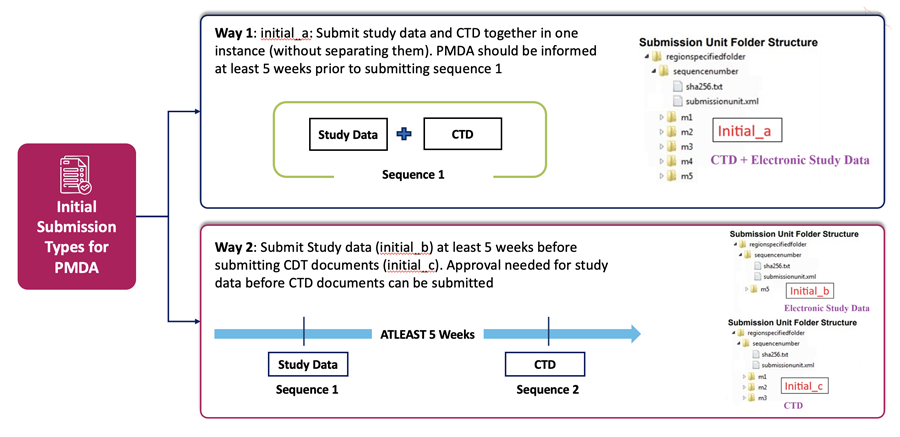
Not only this, but as explained above in the key differences between Japan, US and EU – in Japan we also need to identify the right submission types for Initial NDA submissions and then plan for timelines for submission to PMDA based on the selected category as shown below.
eCTD v4.0 Initial NDA Submission Types in Japan
eCTD Initial NDA submission Types
jp_initial_a
Initial submission unit that covers both the study data and the documents specified by the CTD guidance.
jp_initial_b
Initial submission unit that covers the study data only.
jp_initial_c
Initial submission unit that covers the documents specified by the CTD guidance.
jp_other
This code may be used only when advised to do so by the regional authority in unavoidable circumstances and where none of the other units is appropriate.
PMDA eCTD v4.0: initial submission Type Methods
What Happens to Submissions in Outdated Formats? Can They Be Transitioned to eCTD v4.0?
Post the mandatory implementation date for PMDA and other Health authorities, submission of applications in non eCTD v4.0 format can lead to rejections or requests for re-submission or slower review process.
But the good news is that the eCTD v4.0 structure allows for reusability of documents submitted in previous formats. However, the process and effort involved will depend on several factors:
Transitioning from a paper-based submission or a very old electronic format (like NeeS) will be significantly more complex than transitioning from eCTD 3.2.2.
A well-structured older dossier will be easier to map to the eCTD v4.0 structure.
Specialized software and personnel with expertise in eCTD v4.0 are necessary for a successful transition.
Some Health Authorities may provide guidance or tools to assist with the transition, while others might leave it entirely to the applicant.
Key Steps for Transitioning to eCTD v4.0
The transition of previous submissions will require a thorough knowledge of the eCTD v4.0 and the right process for execution like:
Gap Analysis
Compare the structure and content of the old dossier with the requirements of eCTD v4.0, identifying any gaps in data or format.
Document Reorganization
Reorganize the existing documents to fit the Module structure and the more granular structure of eCTD v4.0.
Metadata Updates
Update the metadata for all documents to comply with the eCTD v4.0 specifications, including controlled vocabularies and new data elements like UUIDs (Universally Unique Identifiers).
XML Generation
Create the new eCTD v4.0 XML backbone file that accurately reflects the dossier structure and links all the documents with their corresponding metadata. eCTD v4.0 uses a single XML file, unlike the multiple XML files in eCTD 3.2.2.
Technical Validation
Validate the newly created eCTD v4.0 submission using appropriate software to ensure technical compliance with the Health Authority's specifications.
Submission
Submit the newly formatted eCTD v4.0 dossier to the Health Authority.
Hence, it’s important to work with the right partner to ensure that eCTD v4.0 submissions can be executed seamlessly, including for older submissions.
Ready to streamline your regulatory submissions? In our next blog, we’ll walk you through some regional differences between controlled vocabularies, need for working with tools compliant with latest mandates and some best practices for a successful submission from Freyr’s experience working with clients and health authorities across Japan, US and EU.
Curious about how we can help you achieve faster, more efficient submissions? Reach out to Freyr today!

About the Authors
Ragavendran Babu (Ragav) is a seasoned regulatory operations leader with over 20 years of experience across global health authority submissions. As Director of Regulatory Operations at Freyr, he specializes in regulatory submission strategy, eCTD/NeeS/Paper submissions, and emerging innovations in regulatory technology, including AI applications and structured content solutions.
Ragav has served as an industry subject matter expert, successfully piloting and implementing evolving regulatory standards such as eCTD v4.0, SPL, SPM, and ePI across major global markets. He works closely with technology teams to align regulatory business needs with practical, scalable solutions, while also leading internal and external training programs to build regulatory readiness.
He holds a Bachelor’s degree in Chemistry and a Master’s in Business Management (Systems & Information Technology).

Dr. Mohit Batra is a medical doctor and an accomplished strategy consultant with over 12 years of global experience in life sciences, digital health, and regulatory affairs. As Senior Director and Global Business and Strategy Head of Regulatory Operations at Freyr Solutions, he brings a unique blend of medical expertise and strategic insight. Dr. Batra holds an MD from Delhi University and an MBA from the Indian School of Business.