Medical Device Registration in Japan - Overview
Japan is the second biggest market for Medical Devices and is expected to reach US $74.4 billion by 2025. Key factors driving growth for medical device registration in Japan are:
- Population increasingly skewed toward geriatrics
- Increase in life-style diseases and chronic health conditions
- A wider population base being covered under universal health insurance
- Regulatory changes to facilitate innovative technologies
Though the market looks promising, the major barrier in obtaining medical device approval in Japan market is the stringent Pharmaceuticals and Medical Devices Agency (PMDA) regulations that companies must abide by. To manage their product registrations and liaise with Japan’s regulatory authorities, all foreign medical device and pharmaceutical companies selling in Japan must assign a licensed Japanese Agent.
Regulatory Authority: Pharmaceuticals and Medical Devices Agency (PMDA) working under the Ministry of Health Labour and Welfare (MHLW).
Regulation: Pharmaceuticals and Medical Devices Act (PMD Act).
Authorized Representative: Marketing Authorization Holder (MAH) or Designated Marketing Authorization Holder(D-MAH).
QMS Requirement: Ordinance No. 169, MDSAP Certification.
Assessment of Technical Data: Medical devices classified as Class II and Class III and for which certification criteria have been established are subject to review/audit by a Certification Accreditation Body (CAB). All other medical devices will be audited by the PMDA.
Validity of License: QMS license and Foreign Manufacturer Registration certificate are valid for five years. After this period, licenses and certificates must be renewed. Moreover, any change in the information requested at the time of device registration may be subject to a partial change, minor change or a new application.
Labelling Requirements: Pharmaceutical and Medical Devices Act - PMD Act.
Language: Japanese.
Japan Medical Device Classification
Japan has a clear-cut device classification system. The devices are classified into 4 classes based on the risk associated with the device. The registration procedure, document requirements, technical assessment varies with the class of the device.
| Device Class | Risk |
| Class I | Low Risk |
| Class II | Low/Medium Risk |
| Class III | Medium/High Risk |
| Class IV | High Risk |
Market Authorization Holder (MAH)
A Medical Device (MAH) refers to an entity responsible for the market within Japan as stipulated by Japan's Pharmaceutical and Medical Devices Act (PMD Act). Specifically, they are accountable for ensuring the safety and quality of medical devices. These entities must obtain formal approval from prefectures and employ personnel who meet qualification requirements and operate their business in accordance with strict regulations and requirements.
The MHLW provides a Special Foreign Approval System (SFAS) for foreign manufacturers of Class II, III, and IV medical devices. This allows them to obtain medical device marketing approval (approval by PMDA) or certification (approval by CAB) under their own name, thereby becoming deemed MAH. Under this system, foreign manufacturers are required to delegate certain tasks to appointed MAH.
Foreign Manufacturer Registration (FMR)
All the foreign manufacturing companies which intend to export their devices into Japan must register themselves with the MHLW. This registration procedure is called as Foreign Manufacturer Registration (FMR), which formerly was known as “Foreign Manufacturer Accreditation (FMA)” or “Accreditation of Foreign Manufacturers (AFM)”. This registration is valid for five years, so a renewal must be required.
Japan Medical Device Registration
The registration pathway in Japan depends on various factors such as class of device, assigned Japanese Medical Device Nomenclature – JMDN, availability of predicate device and availability of associated Japanese Industrial Standard (JIS). Various registration pathways in Japan are detailed below -
- Notification (Todokede - 届出): It applies to general Medical Devices (Class I), wherein manufacturers can file a pre-market submission to the PMDA. This is a notification, and no review/assessment by the PMDA will be conducted.
- Third Party Certification (Ninsho - 認証): It applies toClass II (and a limited number of Class III) devices, for which certification standards have been set. The process is like the European CE Marking process, where reviews are outsourced to a third party like a CAB.
- Approval (Shonin - 承認): Class II and III devices with no specific certification standard and all Class IV devices are subject to the pre-market approval process, also known as Shonin. This application must be submitted to the PMDA and ultimately obtain approval from the MHLW.
Process flow
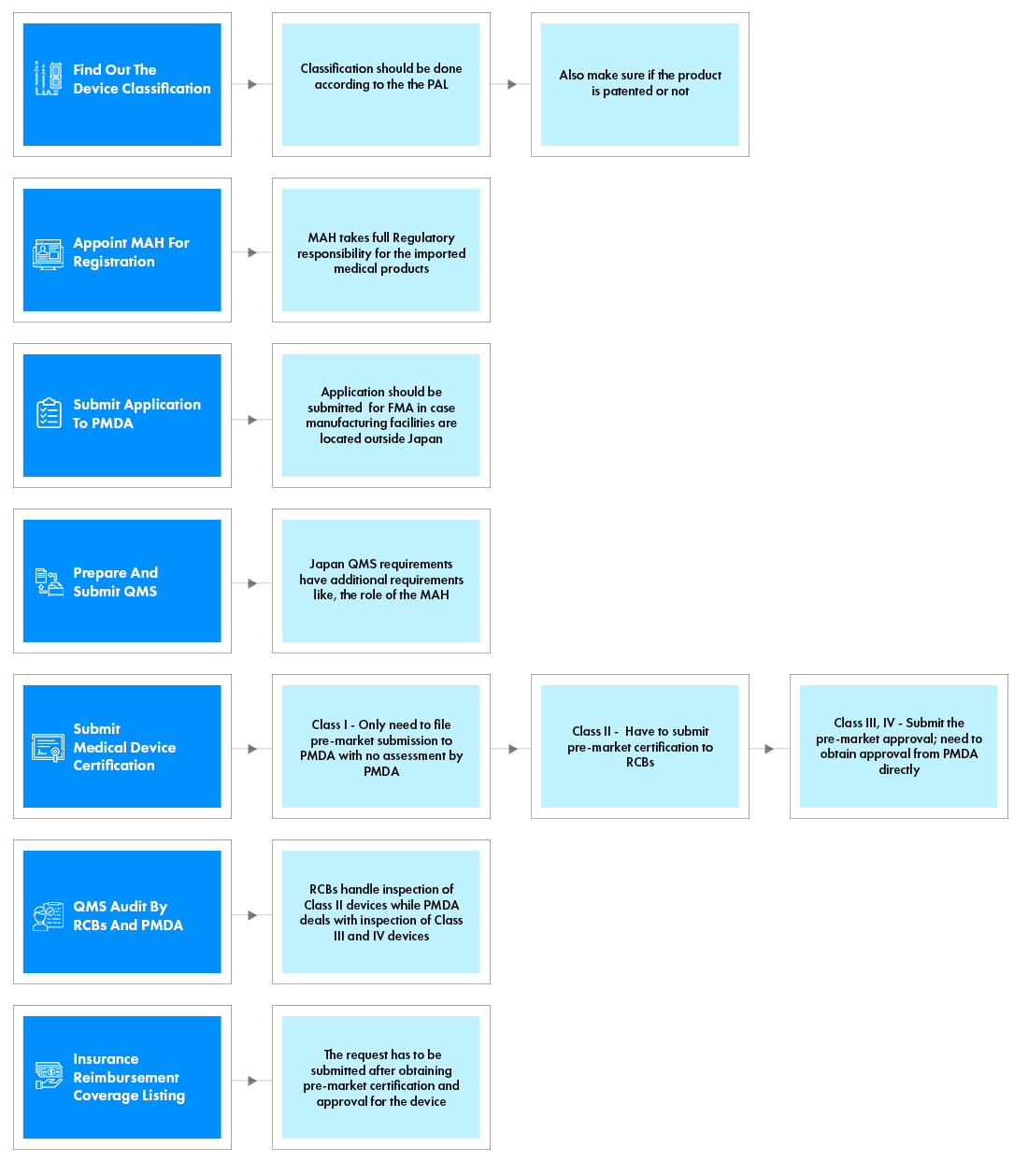
MDV registration process flow
- Find out the device classification/JMDN:
- Classification should be done according to the device classification AND JMDN codes and generic names.
- Also make sure if the product is patented or not.
- Appoint MAH for registration:
- MAH takes full regulatory responsibility for the imported medical products.
- Prepare applications, technical documentation summary and other documents required for the registration route identified:
- Application should be submitted for FMR in case manufacturing facilities are located outside Japan. This is also subject to the device’s sterilization company.
- Make sure that the FMR process has been done and is valid, and that warehouse/distributor arrangements are in place.
- Prepare QMS conformity survey application and required documents:
- Be compliant with Ministerial Ordinance #169.
Having certification of ISO 13485, and/or MDSAP with Japan in scope, would be very beneficial. - The QMS conformity survey also extends to warehouses, which are storage locations for finished products in Japan.
- Be compliant with Ministerial Ordinance #169.
- Make Submissions:
- Class I - Only need to file Notification (Todokede - 届出) documents to PMDA with no assessment by PMDA.
- Class II and III with approval criteria (Ninsho criteria) - Must submit Third Party Certification (Ninsho - 認証) to CABs.
- Class III without Ninsho criteria and IV - Submit to PMDA (Approval (Shonin - 承認)).
- Pay fees for Class II, III and IV.
- Technical documentation review and QMS audit by CABs and/or PMDA:
- Reviews (audits) by the authority to which the application is submitted.
- Additional information may be requested. There will be multiple exchanges of inquiries with the authority.
- Approval
Upon approval (except for Class 1), the Minister of Health, Labour and Welfare or the CAB, will issue a device registration certificate (marketing authorization approval or marketing authorization certificate) and a certificate of compliance with QMS standards.
Post Approval Medical Device Life Cycle Management
Freyr supports the foreign manufacturers in end-to-end Medical Device lifecycle management, including post approval activities, such as:
- Post approval change management - modifications to existing Medical Device approvals such as, addition of new variants, accessories; addition of new indications of use among others.
- Maintenance of approvals and registration through timely payment of administrative and registration fees
- Renewal of licenses
- Liaising between CAB or PMDA and the manufacturer
- Importation Management
Despite having an established medical device Regulatory framework, navigating through Japan’s device classification system and respective registration procedures may require proven expertise.
Freyr, as a strategic Regulatory partner, provides end-to-end Medical Device Regulatory services that span across quality control, classification, clinical safety, and market access. We assist clients in all the procedural challenges right from Regulatory intelligence to dossier preparation and submission to product registration.
Summary
| Controls | Device Class | Risk / Classification Criteria | Approval Routes | Agency | Timelines |
| General | Class I | Low Risk | Pre-Market Submission (Todokede) | NA | 1 Month |
| Specified Controlled | Class II | Low/Medium Risk | Pre-Market Certification (PMC / Ninsho) | Notified Body | 3-5 months |
| Controlled | Class II | Medium Risk | Pre-Market Approvals (PMA / Shonin) | PMDA | 7-9 Months |
| Highly Controlled | Class III | Medium / High Risk | Pre-Market Approvals (PMA / Shonin) | PMDA | 9 - 12 Months |
| Highly Controlled | Class IV | High Risk | Pre-Market Approvals (PMA / Shonin) | PMDA | 13-16 Months |
Freyr Expertise
- Regulatory Intelligence Support
- Regulatory Due Diligence
- Device Registration
- Submission Management to PMDA or notified bodies
- Data reliability inspection
- QMS inspection
- Designated Marketing Authorization Holder (D-MAH)
- Marketing Authorization Holder (MAH)
- Foreign Manufacturer Registration (FMR)
- Reimbursement Application support (National health insurance reimbursement)
- Label Support (Label requirement verification)
- Warehouse/Distributor introduction support
- License transfer
Freyr Advantages
- Able to support multiple regions. Not only Japan medical device registration, but also overseas registration services are available.
- In addition to establishing submission documents, we can provide communication support with overseas agents and manufacturers.